
System Impact Assessment: A Risk Management Framework For A COVID World
By Wai Wong, VP Validation, Pharmatech Associates

The new normal created by the recent COVID-19 pandemic has created disruptions in many sectors of life sciences. From hospital design to drug manufacturing, the industry is stretched thin on resources while still having to move forward with research and development and manufacturing, especially for rapid COVID response targeted solutions with less than the typical quality structure. Some manufacturers that have never operated in the traditional quality management system before are now required to comply with standards in an industry that cannot tolerate poor quality. Infusing risk management is essential to an organization, especially in terms of facility, utility, and equipment qualification. Although qualification is usually the last step before moving to process characterization and validation, integrating a risk management approach is an effective way to ensure qualification activities are focusing on what matters most — that equipment performs as expected.
Qualification is a critical step in the facility and process development program because it is intended to confirm that the equipment possesses the attributes required by the process, has been installed appropriately, and is capable of performing within the specific unit operation to deliver products within specification. Qualification of facilities, utilities, and equipment should not be treated equally, as some systems and subsystems have a much greater potential for impacting product safety and efficacy. The intelligent use of risk management tools can ensure that the qualification focus is concentrated on those components of the process that have the most potential for impact on the form, fit, and function of the product. This moves qualification from being a documentation exercise to being an essential step prior to establishing the process design space and for reducing risk to the process.
Integrating A Risk Management Framework
A risk management framework (RMF) is a structured approach to identify potential threats to a process and to define the strategy for eliminating or minimizing the impact of these risks, as well as the mechanisms to effectively monitor and evaluate the selected remediation. The value of utilizing an RMF is the insight derived from all participants in the risk assessment using a common definition of risk. Many tools are available manage and reduce risk, such as a hazard and operability analysis (HAZOP), which focuses on identifying possible hazards and operability issues in a process, and a failure mode and effects analysis (FMEA), which is often used to capture the inherent risk in a product’s or process’ design, automation, and user experience.
System Impact Assessment
In terms of the facility and equipment qualification process, one powerful RMF tool that can be utilized is the system impact assessment (SIA), which leverages the basic components of an RMF approach, including an agreed-upon ranking table, to evaluate the degree to which equipment must be characterized and qualified to have confidence it will perform as intended. The first step of an SIA is to determine if the system has direct impact, indirect impact, or no impact on product quality. The SIA will evaluate the system’s materials of construction, control capability, management of data, security, and other criteria essential for the system to operate in its manufacturing capacity. It is important to consider the final use and integration of the equipment, especially if it is to be integrated into an automated factory setting as part of a Pharma 4.0 initiative, as the envelope for qualification should consider the system’s impact on the data being acquired. A system in the manufacturing process will have a much higher impact than an ancillary system, such as instrument air that does not have direct contact with the product. Subject matter experts (SMEs), manufacturing, facilities, and quality assurance are key players in evaluating each system’s impact on product quality, which is typically evaluated by utilizing ranking tables that provide parameters that categorizes the impact.
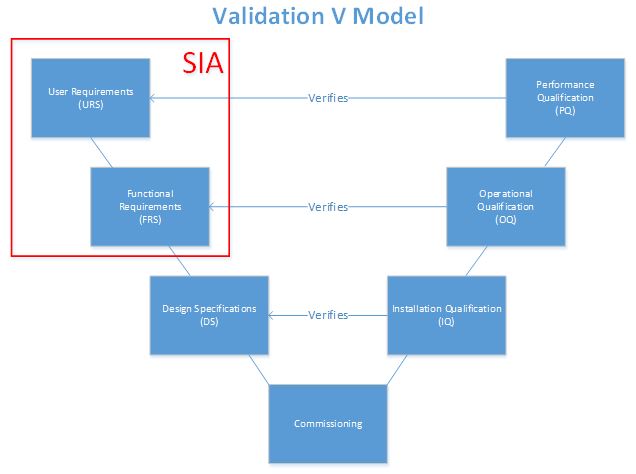
In the past, the classical validation paradigm dictated that all systems required full qualification, regardless of their impact on product quality. The V-model, which does not specifically require an SIA, provided a road map for specification documents that coincided with each qualification protocol. With the addition of the SIA into the V-model, qualification can be limited to systems determined to have direct impact on product quality. As an indirect system may have an impact on a direct-impact system, qualification is not required — commissioning alone is necessary. For example, a reverse osmosis deionized water (RO/DI) system may be classified as an indirect-impact system if the organization uses the RO/DI as only an intermediate to generating water for injection (WFI). The RO/DI has a potential to indirectly affect the product quality when used to produce WFI, a direct-impact system. If, however, the organization uses RO/DI for any purpose that directly affects product quality, it will be considered a direct-impact system.
For companies with the organizational structure to support the initiative, the standards outlined in ASTM E-2500 provide another effective risk-based approach to qualification. ASTM E-2500 utilizes concepts from ICH Q8, ICH Q9, and FDA guidelines for the specification, design, and verification of facilities, utilities, and equipment that have the potential to affect product quality and patient safety. This approach to design, build, and qualification can be combined with an SIA to focus only on elements that have impact to critical process parameters (CPPs) and product critical quality attributes (CQAs). The combination SIA and ASTM E-2500 approach relies on SMEs, typically in the upstream engineering group, to make critical determinations earlier in the equipment selection process, with a focus on performing verification testing at the vendor’s site. This approach redefines roles for quality and technical functions, as it requires understanding both the technical requirements and the quality considerations downstream. Just as with the classical V-model, the SIA is an invaluable tool in this process of identifying direct impact systems that require more rigorous testing and reducing ones that have little or no impact, thereby saving time and costs associated with their qualification.
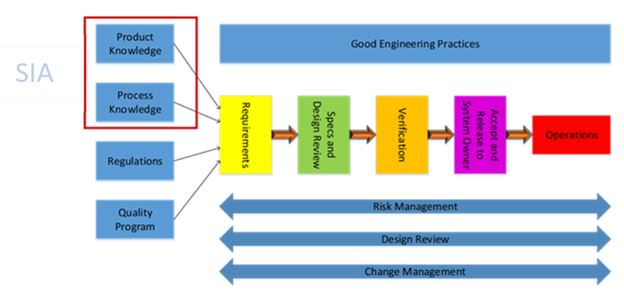
As shown in the figure above, product and process knowledge are crucial in the determination of requirements for the facilities, utilities, and equipment supporting the manufacturing process. ASTM E-2500’s method of earlier definition of requirements refines the specification and design process while also reducing the risk of performing non-value-added work during qualification and validation. The power of the SIA brings out the issues found during the upstream activities, resulting in less costly rework in the downstream process.
Conclusion
In our current COVID pandemic environment, the life sciences industry cannot afford to be sloppy when attempting to meet world needs. Time to market is everything. Beginning or increasing manufacturing capacity should not be performed while running the risk of missing an important issue that has not been properly characterized and analyzed. The key to integrating a risk-based approach should focus on reducing risk to the process and not adding to it. Using management tools such as an SIA to enhance the validation V-model or ASTM E-2500 helps reduce risk, understand capabilities, and what has the biggest impact in the process. In the future, the SIA can be combined with other models of risk management such as digital twins, which create virtual replicas of physical equipment for simulations. Digital twin-type modeling of potential failure modes before testing equipment can provide a lot of value to companies by increasing confidence in both equipment deployment and, consequently, process development. In the case of vaccine development, pairing SIA with digital twin modeling can provide invaluable insight into whether your equipment has the necessary capabilities to accept the process.
About The Author:
 Wai Wong leads the commissioning, qualification, and validation practice for Pharmatech Associates, bringing over 20 years of equipment, facility, and process validation experience in the pharmaceutical, medical device, and biologic industries. He has earned an international reputation for expertise in validation, having demonstrated success in navigating the complex requirements of the FDA, EU, and PIC/S compliance. A Six Sigma Green Belt, he holds a B.A. in molecular and cell biology with emphasis on biochemistry from the University of California at Berkeley. He lives in the San Francisco Bay Area with his wife and children.
Wai Wong leads the commissioning, qualification, and validation practice for Pharmatech Associates, bringing over 20 years of equipment, facility, and process validation experience in the pharmaceutical, medical device, and biologic industries. He has earned an international reputation for expertise in validation, having demonstrated success in navigating the complex requirements of the FDA, EU, and PIC/S compliance. A Six Sigma Green Belt, he holds a B.A. in molecular and cell biology with emphasis on biochemistry from the University of California at Berkeley. He lives in the San Francisco Bay Area with his wife and children.