
Sterility Assurance: The Fundamentals
By Yadnyesh Patel, microbiology subject matter expert

Sterility assurance is a critical aspect of the production of pharmaceuticals, biopharmaceuticals, medical devices, and other sterile products, encompassing all measures taken to ensure the absence of viable microorganisms that could compromise patient safety. This article series outlines the key components of sterility assurance, including the validation of sterilization processes, environmental monitoring, and stringent quality control measures. The significance of sterility assurance extends beyond regulatory compliance, playing a vital role in maintaining product efficacy, protecting public health, and safeguarding the integrity of healthcare interventions. Robust sterility assurance practices not only prevent potentially life-threatening infections but also enhance consumer confidence and brand reputation, contributing to the overall success and sustainability of manufacturing organizations. Continuous advancements in sterilization techniques and testing methodologies further reinforce the importance of sterility assurance in an increasingly complex and demanding global healthcare environment. Sterility assurance is not merely a technical requirement but an ethical imperative in ensuring the safety and efficacy of sterile products.
Let’s Talk Terminologies
The term sterile refers to the condition or state of an object that is free from any viable form of microorganisms. In another words, sterility means the absence of all viable microorganisms (i.e., bacteria, yeast, fungi etc.), including viruses.
In its strictest sense, an item is considered sterile only when it is free from viable microorganisms. However, this definition poses challenges when applied to actual sterile products due to inherent limitations in testing methodologies. Absolute sterility cannot be verified without destructively testing each individual unit. In practice, microbiological safety is achieved through a series of interrelated controls that collectively provide assurance that products are suitable for their intended use. It is these comprehensive controls — rather than results from in-process or finished product testing — that offer meaningful protection against microbiological risks. The process of ensuring the safety of products labeled as sterile is referred to as sterility assurance (as outlined in USP chapter <1211>). This concept underscores the importance of a systematic approach to sterility that prioritizes robust process controls over reliance on testing alone.
Sterility assurance is a critical aspect of manufacturing processes where sterile conditions are essential for safety and efficacy, such as in the production of injectable drugs, surgical instruments, and other sterile medical products.
Establishing An Effective Sterility Assurance Program
Standards like the International Organization for Standardization (ISO) 13485 and ISO 14644 (for cleanrooms) are widely followed in industries to ensure compliance with sterility requirements.
Establishing an effective sterility assurance program necessitates a thorough understanding of the material intended for sterilization. An initial determination must be made to determine the feasibility of terminal sterilization for the material within its primary container. The chosen sterilization process should achieve an optimal balance between conditions that effectively eliminate any potential bioburden present on or within the item, while simultaneously preserving its essential quality attributes. Based on this evaluation, sterility can be attained through either aseptic processing or terminal sterilization. In some cases, employing a strategy that integrates the protective measures of aseptic processing with the lethal effects of terminal sterilization may provide significant advantages. Regardless of the chosen method, it is crucial to establish robust design parameters, operational protocols, process controls, and monitoring systems to ensure confidence in the sterility outcome. The production of sterile products is influenced by a multitude of factors, which must be carefully considered for their potential impact on the final product's sterility.
While the factors illustrated in the accompanying image highlight key influences, it is important to acknowledge that not all relevant elements may be represented.
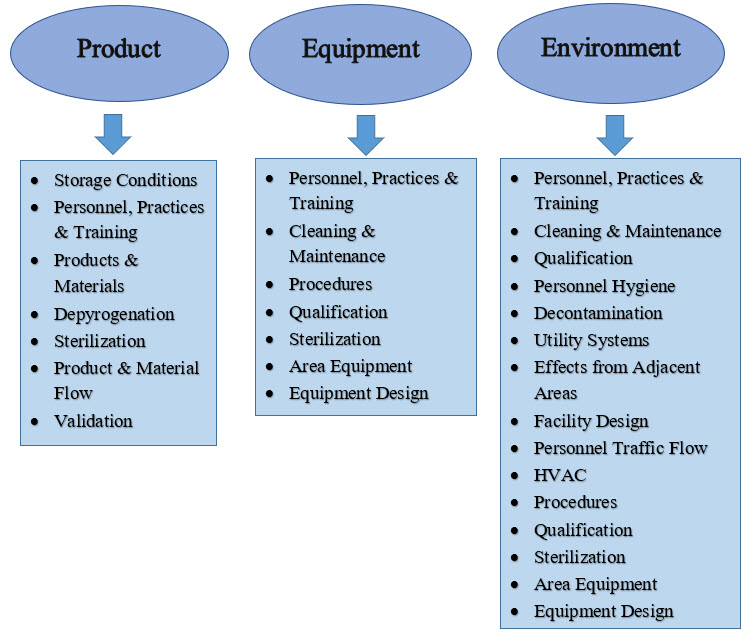
Figure 1: Influences on sterile products.
The decisions made regarding the factors depicted in Figure 1 are critical to the success of the sterility assurance program. The primary objective of process design is to implement contamination controls that mitigate the risk of microbial ingress. This focus is essential whether the process involves aseptic processing or terminal sterilization. Recognizing that operating personnel are the most significant source of microbiological risk informs design choices and operational principles that must be strictly followed in sterile environments. This understanding highlights the necessity of isolating personnel from the aseptic environment and minimizing their interaction with sterilized components and products. To achieve these goals, two complementary practices are essential:
- The use of automation technology to reduce or eliminate personnel interventions and thus personnel-borne contamination.
- The use of separative technologies to eliminate, to the extent technically possible, human sourced contamination.
Thus, implementing appropriate contamination control procedures is paramount in the design and operation of sterile product manufacturing systems.
The design for sterile processing should be driven toward a singular goal of optimizing contamination control with a particular focus on the microbial risk impact of personnel. The specific means vary but should be of prime consideration in process design. The figure below outlines the elements that contribute to sterility assurance.
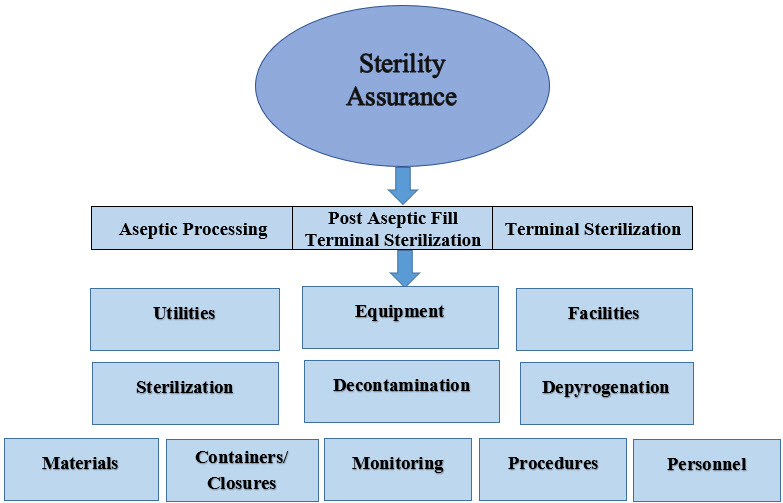
Figure 2: Elements contributing to sterility assurance.
Key Components Of Sterility Assurance
- Sterilization
The most effective means of controlling the microbial population is sterilization, a process that either kills or removes viable microorganisms. In the production of sterile products, sterilization processes are used to prevent microbial contamination. Terminal sterilization processes that reproducibly destroy microorganisms in the final product container are the preferred means for producing sterile products. Sterile products that cannot be terminally sterilized rely on individual sterilization processes (e.g., steam, radiation gas, filtration) for the various materials that comprise an aseptically processed sterile product. In addition, sterilization processes are used for product contact and other non-product contact items used in a variety of applications during the preparation of sterile products to provide absolute control of bioburden.
Aseptic Processing
Techniques that prevent contamination from microorganisms during manufacturing involve maintaining a controlled environment (e.g., cleanrooms), using sterile equipment, and following strict procedural controls.
There are a substantial number of sterile products that cannot be terminally sterilized because of adverse impact on the product/package’s essential properties and therefore must be prepared using aseptic processing. Aseptic processes are designed to prevent the introduction of viable microorganisms into/onto separately sterilized materials during their assembly into a sealed sterile package. Aseptic processes can vary in complexity from comparatively simple filling/sealing to challenging and lengthy manufacturing sequences required for complex items. Regardless of process scale, all of the individually sterilized materials must be protected from contamination from the point of sterilization through closure of the primary package. This is accomplished through adherence to the principles described below in which an ISO 5 condition is maintained when materials are exposed to the environment.
Terminal Sterilization
Terminal sterilization refers to a sterility assurance level (SAL) of 10-6 (SAL6 is considered the standard for injectable and medical devices) and describes the process that ensures that the injectables, medical devices, and implants are sterile at the point of use.
Terminally sterilized products are the lowest-risk category of sterile pharmaceutical products. Unlike products aseptically manufactured under conditions designed to prevent microbial ingress, terminally sterilized products are subjected to a sterilization process that imparts a quantifiable safety level. Terminal sterilization processes achieve this by delivering measurable physical conditions that correspond to microbial lethality.
For terminally sterilized products, sterility assurance is defined in terms of the probability of non-sterility (PNS), or the probability of the terminal sterilization process generating a non-sterile unit (PNSU). Terminal sterilization processes must achieve a consistent validated performance of a PNSU of ≤10 (a probability of not more than [NMT] 1 non-sterile unit in 1 million units produced). The convention by which terminal sterilization cycles are developed and validated ensures that the actual PNSU is typically much lower (better) than the minimum standard of <10.
USP Chapter <1229> summarizes the common requirements for sterilization process design, development, validation, and process control. Terminal sterilization processes share common requirements of well-defined process parameters strictly controlled within defined operating limits. Terminal sterilization must be supported by a system of product disposition, which includes the assessment of critical physical process parameters, pre-sterilization product parameters (e.g., bioburden, container closure integrity), and environmental parameters. Terminal sterilization can rely on parametric release practices to obviate the need for sterility testing.
Post Aseptic Processing Terminal Sterilization
An aseptic process followed by a terminal sterilization process provides superior control over the pre-sterilization bioburden, such that the subsequent sterilization process can be designed with less overall lethality, thereby making it possible to substantially extend the use of terminal sterilization to products with greater sensitivity to the applied energy of the process.
From a patient safety perspective, this approach has the following distinct advantages:
- An adventitious contaminant introduced during aseptic processing is easily killed by the terminal sterilization step, reducing the extent of in-process environmental monitoring performed.
- Bioburden controls for the terminal process are simplified because all units have been aseptically filled.
- Where a product is made using either process alone, the limitations of each (no terminal lethal component in aseptic, more degradation in terminal sterilization) would persist.
- Where bioburden is controlled through aseptic processing, terminal sterilization can be applicable at lower lethality levels.
While product quality attributes can be impaired by “standard” sterilizing conditions, the combined process can utilize less aggressive sterilizing conditions to minimize adverse effects upon the product and primary packaging materials. If the terminal treatment follows aseptic processing, the sterilizing conditions need not be excessive as there is essentially no risk from pre-sterilization bioburden in the filled containers. Sterilization process conditions would be dictated by the specifics of the product in parallel with the establishment of appropriate controls on pre-sterilization bioburden derived from environmental and pre-filtration isolates.
- Sterilization Methods: Various methods, such as steam sterilization (autoclaving), dry heat, gas (ethylene oxide), radiation, filtration, decontamination, and depyrogenation are employed to kill or remove microorganisms. The choice of method depends on the nature of the product and its compatibility with the sterilization process.
- Validation: Sterilization processes must be validated to ensure they are consistently effective. This involves conducting studies to determine the probability of achieving sterility, often referred to as the SAL, typically set at 10-6, meaning there is a one in a million chance of a single viable microorganism being present.
- Environmental Monitoring: Regular monitoring of the manufacturing environment is crucial to detect potential contamination sources. This includes air quality testing, surface sampling, and personnel monitoring.
- Control of Materials: Raw materials, packaging, and components must be handled and processed in a way that prevents contamination. This may involve sterilizing materials before use or ensuring they are produced in a sterile environment.
- Personnel Training and Hygiene: Proper training of personnel in aseptic techniques and the use of personal protective equipment (PPE) are vital to maintain sterility. Personnel hygiene, including the use of sterilized clothing and restricted access to cleanrooms, is also critical.
- Sterility Testing: Final products are often subjected to sterility testing to confirm that they meet sterility requirements. This testing is done in a controlled environment to avoid false positives or negatives.
- Documentation and Compliance: Detailed documentation of all processes, including deviations and corrective actions, is required to demonstrate compliance with regulatory standards (e.g., FDA, EMA). Audits and inspections are conducted to ensure that sterility assurance practices are followed consistently.
These principles collectively ensure that the risk of contamination is minimized and that products meet the necessary sterility standards to ensure patient safety.
In my next article in this series on sterility assurance, I’ll discuss key contributing factors for maintaining sterility assurance. In my last article of the series, I’ll discuss sterility assurance testing methods.
About The Author:
 Yadnyesh Patel earned his master’s degree in microbiology from KBCNM University, Maharashtra, India, in 2009. With over 13 years of extensive experience in quality functions, he has spent time working at pharmaceutical organizations such as Claris Otsuka Ltd, Sun Pharmaceutical Industries Ltd., and Zydus Life Sciences Ltd. Currently, he spearheads microbiological quality functions, document management, audit, and compliance. He possesses substantial expertise in quality management systems, SOPs, documentation management, microbiological test method validations, sterility assurance, aseptic process simulation, and computer system validation.
Yadnyesh Patel earned his master’s degree in microbiology from KBCNM University, Maharashtra, India, in 2009. With over 13 years of extensive experience in quality functions, he has spent time working at pharmaceutical organizations such as Claris Otsuka Ltd, Sun Pharmaceutical Industries Ltd., and Zydus Life Sciences Ltd. Currently, he spearheads microbiological quality functions, document management, audit, and compliance. He possesses substantial expertise in quality management systems, SOPs, documentation management, microbiological test method validations, sterility assurance, aseptic process simulation, and computer system validation.