
Sampling And Testing Of ATMPs: Gaining Insights From PIC/S Annex 2A
By Herman F. Bozenhardt and Erich H. Bozenhardt
 With minimal specific guidance on commercial manufacturing from the FDA and a lack of robust platform commercial processes, some of the finer details of cell and gene therapy manufacturing can make operating companies feel like they are walking on a precarious limb. The majority of the commercial processes have taken a “one off” approach involving risk assessment and discussions with the FDA. In light of the new pipeline of products, the Pharmaceutical Inspection Co-operation Scheme (PIC/S) has developed draft guidance to help point the way. Today that draft guidance is still pointing toward risk assessing certain aspects of manufacturing to get to a new paradigm. One of those aspects is sampling and testing.
With minimal specific guidance on commercial manufacturing from the FDA and a lack of robust platform commercial processes, some of the finer details of cell and gene therapy manufacturing can make operating companies feel like they are walking on a precarious limb. The majority of the commercial processes have taken a “one off” approach involving risk assessment and discussions with the FDA. In light of the new pipeline of products, the Pharmaceutical Inspection Co-operation Scheme (PIC/S) has developed draft guidance to help point the way. Today that draft guidance is still pointing toward risk assessing certain aspects of manufacturing to get to a new paradigm. One of those aspects is sampling and testing.
As we leverage existing knowledge and methods from other biotechnology products, we need to keep the particulars of the specific advanced therapy medicinal product (ATMP) in mind. Taking a sample plan from a vaccine or monoclonal antibody process and applying it to a gene therapy process might be a good place to start because of the similar unit operations. However, most gene therapies are manufactured at a scale (<1 L drug substance) such that sampling at the traditional level would result in significant yield loss. For example, it is typical to take bioburden samples pre and post each unit operation. For a gene therapy process targeting 1 L of drug substance and three purification unit operations, bioburden samples could be 30 percent of the batch.
The draft PIC/S Annex 2A: Manufacture of Advanced Therapy Medicinal Products for Human Use provides an opportunity rationalize sampling:
6.3 Samples should be representative of the batch of materials or products from which they are taken. Other samples may also be taken to monitor the most stressed part of a process (e.g., beginning or end of a process). The sampling plan used should be appropriately justified and based on a risk management approach. Certain types of cells (e.g., autologous cells used in ATMPs) may be available in limited quantities and, where allowed in the MA [Marketing Authorisation] or CTA, [Clinical Trial Authorisation] a modified testing and sample retention strategy may be developed and documented. (Replaces PICS GMP Guide Part I Section 6.12)
Our focus should be to think through how the data is used:
- Is an in-process test used to determine an end point?
- What does a test that is repeated multiple times throughout the process tell us?
Below is an example gene therapy process where an assessment was carried out to reduce the volumes lost to sampling.
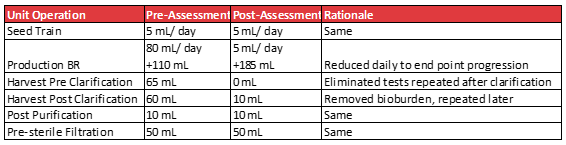
Once the sampling times and tests have been rationalized, the practice of redundant samples should be considered. Annex 2A from PIC/S addresses this topic.
6.5 As a general principle, a reference sample should be of sufficient size to permit the carrying out on at least two occasions of the full analytical controls on the batch foreseen in the CTA or MA. However, it is acknowledged that this may not always be feasible due to scarcity of the materials or limited size of the batches (e.g., autologous products, allogeneic products in a matched donor scenario, products for ultra-rare diseases, and products for use in first-in-man clinical trial with a very small-scale production).
An example of reduced sample volume would be a CAR-T process where routine cell viability and counts are done throughout the culture to determine the end point. Redundant sample volumes would have a major impact on the yield and/or process time due to the small volume of the process (typically <1 L) and frequency of the sample.
Some ATMPs utilize donor derived starting materials. Traditional sample retention policies can be problematic for these types of materials. As an example, long-term storage of blood collections is known to lead to a loss in cell viability due to exposure, thus marginalizing the ability to use the retained sample in a potential future investigation. In addition, for CAR-T treatments, the ability to collect sufficient leukocytes can be difficult for patients who have undergone several rounds of chemotherapy. In this case, an assessment should be made whether retention samples of the donation should be taken at any level. The regulating authorities are understanding these challenges, as evidenced in the sections below from PIC/S Annex 2A.
6.6 Samples of the starting materials should generally be kept for two years after the batch release. However, it is acknowledged that the retention of samples may be challenging due to scarcity of the materials. Due to this intrinsic limitation, it is justified not to keep reference samples of the cells/tissues used as starting materials in the case of autologous ATMPs and certain allogeneic ATMPs (matched donor scenario). In other cases, where the scarcity of the materials is also a concern, the sampling strategy may be adapted provided that this is justified and appropriate mitigation measures are implemented.
6.7 A sample of a fully packaged unit (retention sample) should be kept per batch for at least two years after the expiry date. A retention sample is, however, not expected in the case of autologous products or allogeneic products in a matched donor scenario as the unit produced with the patient’s tissues/cells constitutes what should be administered to the patient. When it is not possible to keep a retention sample, photographs or copies of the label are acceptable for inclusion in the batch records.
6.8 The retention period of samples of starting materials, active substance and intermediate product should be adapted to the stability and shelf-life of the product and, therefore, shorter periods may be justified. In cases of short shelf-life, the manufacturer should consider if the retention of the sample under conditions that prolong the shelf-life (such as cryopreservation) is representative for the intended purpose. For instance, cryopreservation of fresh-cells may render the sample inadequate for characterisation purposes but the sample may be adequate for sterility or viral safety controls (the volume of the samples can be reduced according to the intended purpose). When the cryostorage of a sample is considered inadequate for the intended purpose, the manufacturer should consider alternative approaches (e.g., sample of intermediate product such as differentiated cells).
Consideration of the time between manufacturing and administration is needed. Similarity can be drawn between the stability and handling of live virus vaccines and gene therapies; however, for cell therapies, stability drops significantly with time, and patients may have a timely need for the therapy. In these critical cases, the entire chain of custody and the associated transactions must be modeled completely and realistically. A model method should include all the variabilities, and considerations should include queuing theory and stochastic systems. One of the time constraints can be waiting for final release test results.
6.10 … Where end product tests are not available due to their short shelf life, alternative methods of obtaining equivalent data to permit batch certification should be considered (e.g., rapid microbiological methods). The procedure for batch certification and release may be carried out in two or more stages:
(a) Assessment by designated person(s) of batch processing records, results from environmental monitoring (where available) which should cover production conditions, all deviations from normal procedures and the available analytical results for review in preparation for the initial certification by the Authorised Person.
(b) Assessment of the final analytical tests and other information available for final certification by the Authorised Person. A procedure should be in place to describe the measures to be taken (including liaison with clinical staff) where out of specification test results are obtained. Such events should be fully investigated and the relevant corrective and preventive actions taken to prevent recurrence documented.
To deliver the product in a timely manner and maintain quality assurance, a cell therapy company developed a rapid virus quantification method to enable a high degree of assurance that the product wasn’t contaminated during processing. This, coupled with rapid sterility testing and batch record review by exception, enabled rapid release.
With donor derived materials and processes that are rushing innovative lifesaving treatments to the market, we need to consider the implications of out of specification results. Like a number of other innovative therapies, the therapies can be effective at a wide range of dosage. Efforts must be made to understand the levels of effectiveness versus specifications that focus on safety or stability. Ultimately, specifications must have flexibility and greater range. Most allogenic therapies would be similar to other biotherapeutics, where a strategy of safety stock and additional production runs can mitigate a failure. However, critical care autologous therapies need a different treatment. From PIC/S Annex 2A:
5.46 Where authorised by national law, the administration of a product that does not meet the release specification, might be performed in exceptional circumstances (such as when there is no alternative treatment available that would provide the same therapeutic outcome and the administration of the failed products could be lifesaving).
Given the grave implications to patients, one of the companies with an approved CAR-T has presented its high-level approach to out of specification results. The applicability of national laws has led the company to have different mechanisms for the U.S. and the EU. In the U.S., it has two paths for releasing out of specification product. It maintains an open clinical trial, and if the product is out of commercial release specifications but within the wider limits of the trial, the patients are immediately enrolled in the trial. If the test results are outside the limits of the trial, then the company works with the physician to provide the product under compassionate use guidelines.
While the guidance for ATMPs is not as extensive or specific as traditional therapies, there are ways within the regulatory framework to come to rational and appropriate sampling and testing plans. The challenge is that for new innovative therapies in the ATMP space, cookie cutter or rote guidance will not work. Documented assessments and open communication with the regulators are critical for these unique therapies.
As part of that communication with the regulators, we encourage you to respond to the PIC/S call for comments on revisions to Annex 2.
About The Authors:
 Herman Bozenhardt has 43 years of experience in pharmaceutical, biotechnology, and medical device manufacturing, engineering, and compliance. He is a recognized expert in the area of aseptic filling facilities and systems and has extensive experience in the manufacture of therapeutic biologicals and vaccines. His current consulting work focuses on the areas of aseptic systems, biological manufacturing, and automation/computer systems. He has a B.S. in chemical engineering and an M.S. in system engineering, both from the Polytechnic Institute of Brooklyn. He can be reached via email at hermanbozenhardt@gmail.com and on LinkedIn.
Herman Bozenhardt has 43 years of experience in pharmaceutical, biotechnology, and medical device manufacturing, engineering, and compliance. He is a recognized expert in the area of aseptic filling facilities and systems and has extensive experience in the manufacture of therapeutic biologicals and vaccines. His current consulting work focuses on the areas of aseptic systems, biological manufacturing, and automation/computer systems. He has a B.S. in chemical engineering and an M.S. in system engineering, both from the Polytechnic Institute of Brooklyn. He can be reached via email at hermanbozenhardt@gmail.com and on LinkedIn.
 Erich Bozenhardt, PE, is the process manager for IPS-Integrated Project Services’ process group in Raleigh, NC. He has 13 years of experience in the biotechnology and aseptic processing business and has led several biological manufacturing projects, including cell therapies, mammalian cell culture, and novel delivery systems. He has a B.S. in chemical engineering and an MBA, both from the University of Delaware. He can be reached at via email at ebozenhardt@ipsdb.com and on LinkedIn.
Erich Bozenhardt, PE, is the process manager for IPS-Integrated Project Services’ process group in Raleigh, NC. He has 13 years of experience in the biotechnology and aseptic processing business and has led several biological manufacturing projects, including cell therapies, mammalian cell culture, and novel delivery systems. He has a B.S. in chemical engineering and an MBA, both from the University of Delaware. He can be reached at via email at ebozenhardt@ipsdb.com and on LinkedIn.