
Computer Systems Validation Pitfalls, Part 1: Methodology Violations
By Allan Marinelli and Abhijit Menon

When work is not performed according to protocol instructions — by cutting corners on quality outputs with the focus on profit as the main driver or by creating unnecessary work that reduces efficiency — this can result in pharmaceutical, medical device, cell & gene therapy, and vaccine companies losing profits, efficiencies, and effectiveness. The reason is a lack of QA leadership management/technical experience, among other poor management practices, coupled with the attitude that “Documentation is only documentation; get it done fast and let the regulatory inspectors catch us if they can to find those quality deficiencies during phase 1 of the capital project and our validation engineering clerks will correct them later”. Consequently, these attitudes can lead to at least 15 identified quality deficient performance output cases during the validation phases, as will be discussed in this four-part article series.
Rather than focusing on the root causes as a result of lack of effectiveness/efficiencies — such as when Company ABC indicated to the FDA that the entire phase of the project will be entirely re-executed as part of phase 2 — a large proportion of companies ruined by poor management will often blame the design/validation engineers who attempted to make a difference when:
- executing validation protocols pertaining to upstream/downstream manufacturing equipment (e.g., DeltaV Manufacturing Execution System (MES), bioreactors, centrifuges, autoclaves, depyrogenation ovens, water for injection generators/distribution, bionic medical devices, radio frequency devices, etc.) or
- upon executing any laboratory computerized systems (e.g., nuclear magnetic resonance, gas-chromatography mass spectrometry, inductively coupled plasma mass spectrometry, laboratory information management systems, cloud-based systems, etc.) or
- upon executing any electronic documentation systems/information technology systems (e.g., artificial intelligence driven software for quality management systems, electronic batch records, enterprise resource planning software system, SaaS, Azure platforms, etc.).
Poor management can be defined as follows:
“Poor management means having a negative impact on employees and the company. Instead of leading them to success, a poor manager holds them back. Now, poor management can take many different forms. However, they all result in low-functioning teams. And they all involve failing to put people first."1
There are two main levels of classifications, namely, individual level or company level, that can represent poor management:
Individual Level 1
This includes, for example, not giving credit to employees/consultants; micromanaging; staying hands-off; having poor emotional intelligence; being too negative; not communicating clearly; being disorganized; not being equally inclusive of everyone in the team; neglecting employees’ growth; putting too much pressure on employees; not taking feedback well; not being fueled by their passion and values; ignoring wellness; lack of cGMP training/technical training where warranted; being disrespectful; or a combination of these factors.
Company Level 1
These include, for example, not being agile enough to jump on trends; harping on problems while missing what’s going well; ignoring real problems affecting success; the culture is suffering, as company leadership isn’t setting a strong example; employees/consultants aren’t building their skills; people are disengaged despite metrics artificially showing that people are engaged.
In part one of this series, we will not go into further detail about the implications or ramifications of poor management practices, but we will delineate the resultant quality deficiency/performance output cases during the execution of validation protocols among the pharmaceutical, biopharmaceutical, vaccine, medical device, and cell/gene therapy industries with regard to violation of adherence to the methodology section of the protocol.
Violation Of Adherence To The Methodology Section Of The Protocol
We’ll use a real case study to illustrate the concepts below.
As part of the “methodology section” of the preapproved validation protocol for Equipment A/System A, the instructions indicate that during the execution phase the validation engineer is to provide the required evidence of testing intrinsic to Attachments 1, 2, 3, etc., such as software screenshots, raw data calculations, or any other applicable evidence requested in the instruction of each test step, with an accompanying nomenclature identification as indicated.
NOTE: Each page of the accompanying attachment during the execution of the protocol/script must be identified using an X of Y format in which the X represents the current page while the Y represents the total number of pages while identifying for additional traceability (meta data) the protocol number, attachment number, test section/case, test-step, and initial/date of the validation engineer.
- For attachments of five pages or fewer: Identify each page.
- For attachments of more than five pages: Identify the first page and the last page.
Observation: It was noticed during reviews of the post-execution phase that a large proportion of the validation engineers were inconsistently identifying the attachments within the same family of the executed Equipment A/System A, which directly represents a violation of the “Adherence to the Methodology” section of the protocol.
Moreover, a huge percentage of the already executed protocols involving many different types of equipment/systems performed by various departments exhibited these violations, which were previously approved by numerous rubber-stamping QAs (even at the senior QA level) and accompanied by other parties.
See two different examples representing this violation below.
Example 1 — Execution of Protocol/Script (missing identification of the last 200 pages of the accompanied labeling of the attachment as part of the evidence of the executed protocol/script)
Some validation engineers were only identifying the first page of the supporting attachment, because that is how they performed the execution in previous companies. This is coupled with the fact that the poor management of Company ABC specifically instructed the validation engineers to get it done “fast” without providing ample hands-on technical training. Company ABC assumed that by artificially completing the theoretical matrix training assigned by the training department prior to the execution phase, this would provide the recipients with sufficient training to perform their job scope. Management did not realize that many recipients from various departments, including validation engineers, confirmed that the online trainings were completed by simply consecutively clicking screens without even reading/understanding what is being presented, including the training list of SOPs/WIs, protocol directions, methodologies, etc.
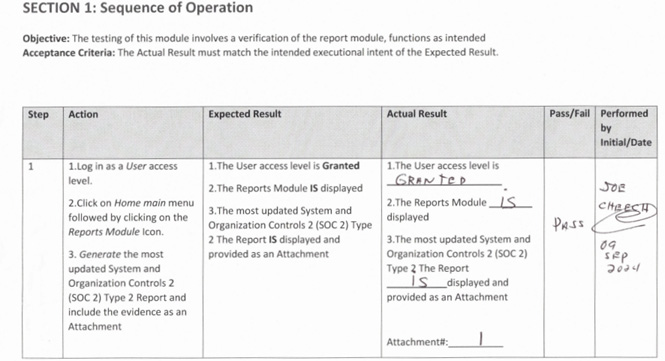


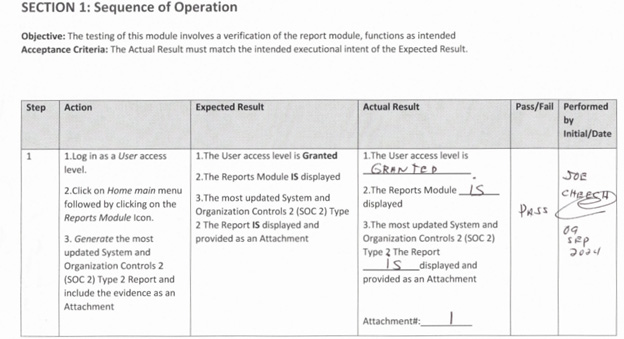
Example 2 — (Missing full meta data identification of the accompanied labeling of the attachment as part of the evidence of the executed protocol/script)
Some validation engineers were only identifying the page numbers, without identifying the associated protocol number, test section, and relevant test while not being aware of the ALCOA+++ principles and updated GAMP 5 Edition 2.


Recommendation/Potential Solution
We recommend that each stakeholder involved, with respect to their assigned roles, when reviewing and approving any executable validation protocols, always read, understand, and adhere to all of the technical instructions, requirements, and contents specified in the protocols prior to performing any validations, without cutting corners on quality while meeting industry requirements.
Moreover, by clearly understanding and carefully being trained on the project scopes at the inception phase of any project, the projects will be better and smoothly executed once the stakeholders, including QA lead/management, fully understand all of the accompanied goals/drivers. This will further ensure that reworks (e.g., repeating entire or partial phases) of any projects, including any of respective implementation of any kind/type of products or services, are avoided altogether.
One can ask, if there is one easily observed violation involving not adhering to the instructions of the protocol requirements, how many other violations have yet to be found that can easily be found by experienced regulatory inspectors?
Conclusion
Always do it right the first time by paying attention to the details stipulated in the protocol, rather than repeating the tests, protocols, and validations that could have easily been avoided at the inception of the capital project. This would allow additional time to focus on other matters like performance troubleshooting, continuous improvements, adding validated data driven decision tools (e.g., validated artificial intelligence-driven software data tools), etc., toward a more robust, efficient, effective, and successful execution and further saving cost on expensive capital projects.
In part 2, we will examine quality deficiencies/performance output cases including unnecessary “nice to haves” (wasting execution efficiencies while not putting enough time where it matters):
- Not clearly defining the “family approach”
- Confusion interpreting calibration versus certified records and material being used to document in one attachment
- Previous QAs’ misinterpretation of protocols’ directives and proposing “nice to haves” that unnecessarily increase execution times (bottom-line: reducing the execution efficiencies and effectiveness)
- Misleading deviation identifications (numerous protocol identifications of the same deviation)
- Executions incomplete (status have not been completely reviewed and populated by the validation department) prior to submitting to QA
Reference
1. Poor Management: 21 Problems & How to Solve Them - Primalogik
About The Authors:
 Allan Marinelli is the president of Quality Validation 360 Incorporated and has more than 25 years of experience within the pharmaceutical, medical device (Class 3), vaccine, and food/beverage industries. His cGMP experience has cultivated expertise from manufacturing/laboratory computerized systems validations, computer software assurance, information technology validation, quality assurance, engineering/operational systems validation, compliance, remediation, and other GxP validation roles controlled under FDA, EMA, and international regulations. His experience includes commissioning qualification validation (CQV); CAPA; change control; QA deviation; equipment, process, cleaning, and computer systems for both manufacturing and laboratory validations; artificial intelligence (AI)-driven embedded software and machine learning (ML); quality management systems; quality assurance management; project management; and strategies using the ASTM-E2500, GAMP 5 Edition 2, and ICH Q9 approaches. Marinelli has contributed to ISPE baseline GAMP and engineering manuals by providing comments/suggestions prior to ISPE formal publications.
Allan Marinelli is the president of Quality Validation 360 Incorporated and has more than 25 years of experience within the pharmaceutical, medical device (Class 3), vaccine, and food/beverage industries. His cGMP experience has cultivated expertise from manufacturing/laboratory computerized systems validations, computer software assurance, information technology validation, quality assurance, engineering/operational systems validation, compliance, remediation, and other GxP validation roles controlled under FDA, EMA, and international regulations. His experience includes commissioning qualification validation (CQV); CAPA; change control; QA deviation; equipment, process, cleaning, and computer systems for both manufacturing and laboratory validations; artificial intelligence (AI)-driven embedded software and machine learning (ML); quality management systems; quality assurance management; project management; and strategies using the ASTM-E2500, GAMP 5 Edition 2, and ICH Q9 approaches. Marinelli has contributed to ISPE baseline GAMP and engineering manuals by providing comments/suggestions prior to ISPE formal publications.
Abhijit Menon is a professional leader and manager in senior technology/computerized systems validation while consulting in industries of healthcare pharma/biotech, life sciences, medical devices, and regulated industries in alignment with computer software assurance methodologies for production and quality systems. He has demonstrated his experience in performing all facets of testing and validation including end-to-end integration testing, manual testing, automation testing, GUI testing, web testing, regression testing, user acceptance testing, functional testing, and unit testing as well as designing, drafting, reviewing, and approving change controls, project plans, and all of the accompanied software development lifecycle (SDLC) requirements.
Check out the other articles in this series: