
2023–2024 Trends In FDA Form 483s For Pharmaceutical Formulation Facilities
By Ajay Pazhayattil, Satish Joshi, and Marzena Ingram

Insights from FDA inspection trends serve as valuable resources for understanding regulatory focus areas and identifying potential vulnerabilities within quality systems. The publicly available inspection observation data provide a strong rationale for immediately addressing many of the challenges faced by manufacturers of FDA-regulated drug products. By leveraging these insights, organizations can better understand how various factors influence compliance and prepare more effectively for future inspections. FDA Form 483 is issued to a firm’s management at the conclusion of an inspection when an investigator(s) has observed any conditions that, in their judgment, may constitute violations of the Food, Drug, and Cosmetic (FD&C) Act and related requirements. FDA investigators ensure that each observation on Form 483 is clear, specific, and significant, providing a robust assessment that reflects the site's efforts in maintaining regulatory standards.1 This is a continuation of the Outsourced Pharma article2 published on June 29, 2023.
Methodology
A comprehensive review of observations from drug facilities sheds light on the state of compliance and regulatory practices in the pharmaceutical industry. Form 483s issued during the 17-month period from Jan. 1, 2023, to May 31, 2024, were considered for the analysis. The following U.S. FDA data sets were used for the analysis:
- Inspections data set3
- Citation data set
- Published 483s data set
- FDA drug establishment annual registration status4
- FDA warning letter database5
Only currently active drug product formulation facility sites per current drug establishment annual registration status were considered in the analysis. This includes brand, generic, and CDMO solid oral, semi-solid, and injectable manufacturing facilities. Manually prepared 483 forms are excluded from this assessment. The agency updates these data sets weekly, ensuring that the findings reflect the most recent inspections and observations. The assessment does not include stand-alone service providers such as analytical labs, sterilization, relabeling, repackage, particle size reduction, positron emission tomography drug production, gas, compounding pharmacies, and API manufacturing facilities. The analysis deduced demographic patterns and U.S. FDA inspection trends based on the available data for the period.
Inspection Classification (2023–24)
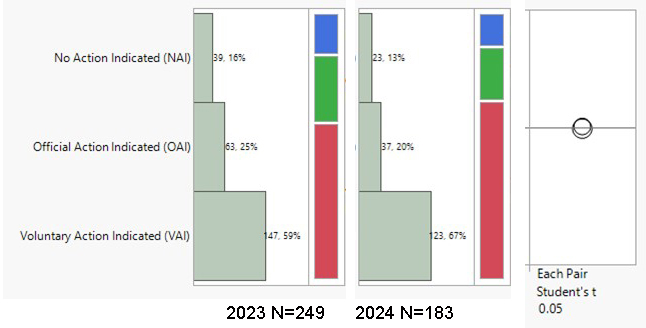
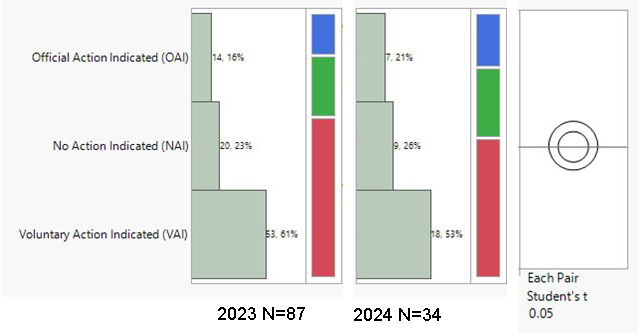
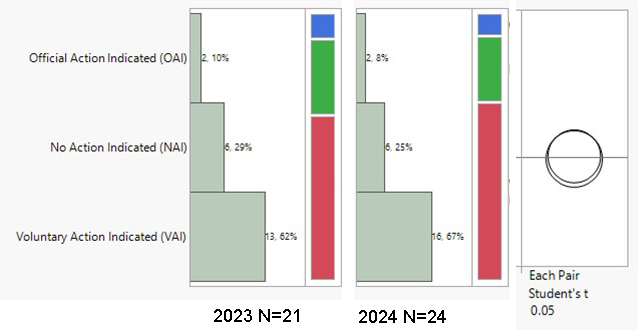
During the period from 2023 to the end of May 2024, the highest volumes of U.S. FDA inspection 483s issued for finished formulation-sites were in the U.S. (59.5% of total 483s issued), India (10.4%), and Canada (8.7%). The inspection classifications — Official Action Indicated (OAI), Voluntary Action Indicated (VAI), and No Action Indicated (NAI) — for the period were analyzed. A notable overall trend is the increase in OAI inspection outcomes and the decrease in NAI status. There was a 5% decrease in OAI outcomes in the U.S., with VAI outcomes remaining high at 67%. Conversely, in India, there was a concerning 5% increase in OAI outcomes. During the period 2023–end of May 2024, 45% of the total OAI status issued to foreign sites was in India, followed by 11% in South Korea and 9% in Canada. In the previous year (2022–May 2023), Indian sites also accounted for 45% of the foreign site OAI status, with 14% in Mexico and 8% in Canada.
Drug Product Facility Inspections and Outcomes in % (NAI, VAI, OAI)

Click on the image above to enlarge.
USA
India
Canada
The choropleth map vividly illustrates a demographic divide in the distribution of U.S. FDA inspections. In the United States, the number of domestic inspections aligns closely with the high volume of pharmaceutical formulations produced, reflecting a regulatory presence proportional to manufacturing output. In India, there is a notable need for a steady increase in inspections. A recent announcement6 highlights the FDA's strategic focus on addressing the region's growing manufacturing volumes, ensuring expanded oversight to maintain compliance and quality across pharmaceutical facilities. This geographic emphasis underscores the FDA's ongoing adaptive approach to the dynamic landscape of global pharmaceutical manufacturing.
Choropleth Map
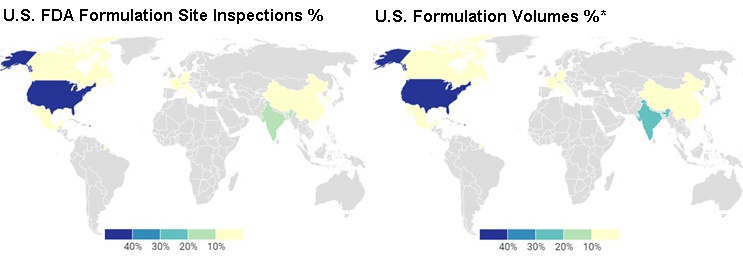
*Source: The geography of prescription pharmaceuticals supplied to the U.S.: levels, trends, and implications.7 This is an older data set; the estimate is that the finished formulation volumes from India have increased with the higher number of ANDA approvals in recent years.
Inspection Observations
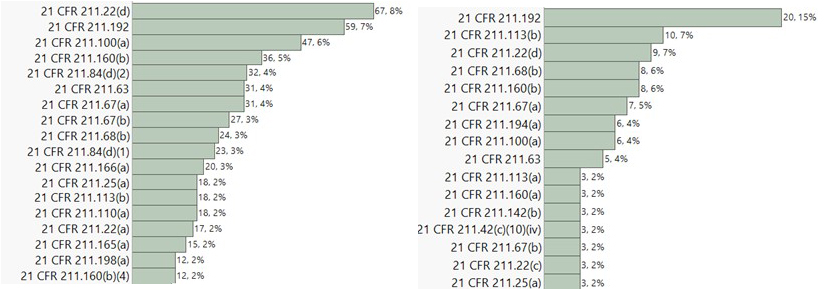
Inspections of formulation facilities in the U.S., India, and Canada during the period primarily led to the observations associated with the below listed CFR sections:
USA India
Canada
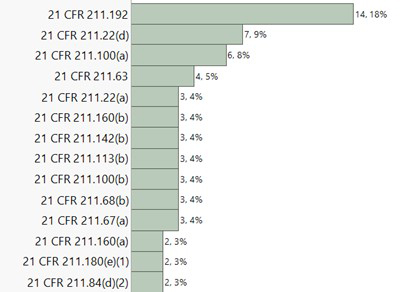
Observations related to 21 CFR 211 sections 22(d), 192, and 100(a) were consistently observed to be high across all three major inspection regions. Notably, observations associated with 21 CFR 211 section 113(b) were most prevalent in India. Section 113(b) pertains to having appropriate written procedures designed to prevent objectionable microorganisms in drug products not required to be sterile, which include validation of all aseptic and sterilization processes. CFR 211 section 22(d) is associated with the responsibilities and procedures applicable to the quality control unit. CFR 211 section 192 primarily focuses on investigation robustness. It requires all drug product production and control records to be reviewed and approved by the quality unit to determine compliance with all established, approved written procedures before a batch is released or distributed. Any unexplained discrepancy or the failure of a batch or any of its components to meet any of its specifications should be thoroughly investigated. The investigation should extend to other batches of the same drug product and other drug products that may have been associated with the specific failure or discrepancy. CFR 211 section 100(a) states that written procedures for production and process control are to be designed by the SMEs and approved by the quality unit to assure that the drug products have the SISPQ they represent to possess. A correlational analysis was subsequently performed across the top 10 483 observation categories for further insights. The correlation probability is listed below:
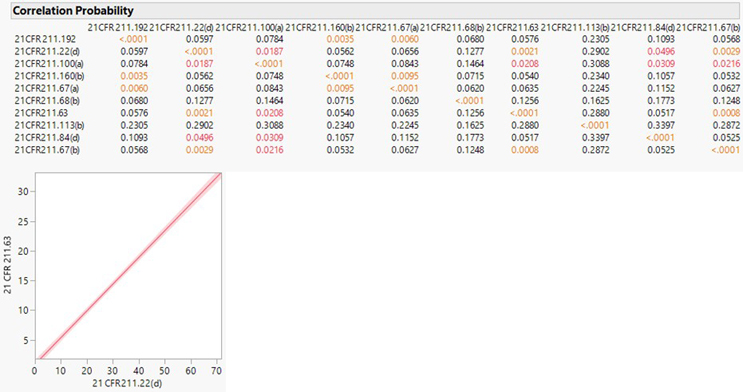
A definite positive correlation was observed between 21 CFR 211.22(d) (responsibilities and procedures applicable to the quality unit) related observations and 21 CFR 211.63 (equipment used to be of appropriate design, adequate size, and suitably located to facilitate operations for its intended use and its cleaning and maintenance) related observations.
What Can We Infer From This Data?
The observed decrease in OAI outcomes for domestic U.S. facilities reflects the effectiveness of regulatory actions, such as warning letters and subsequent remediation efforts involving subject matter experts (SMEs). In contrast, the increase in OAI outcomes at Indian facilities underscores the need for immediate proactive implementation of robust remedial measures, focusing on the top areas of current regulatory concern, i.e., investigation effectiveness, validation and process robustness, and contamination control. The FDA has planned for a steady increase in inspections for drug manufacturing units in India, reflecting the necessity for heightened oversight in response to the region's growing manufacturing volumes for the U.S. market. For the review period, the regulator's primary concerns during domestic and global inspections were on ensuring:
- responsible quality management,
- robustness of investigations, and
- ensuring stringent production and process controls (process validation).
In addition, Indian formulation facilities faced several observations related to contamination control and validation/controls on sterile manufacturing processes. The trend may suggest the regulator's lack of confidence in older aseptic facilities that have been delaying the adoption of the latest containment technologies essential for enhanced sterility assurance, critical for patient safety. In a recent warning letter, the U.S. FDA emphasized the ineffectiveness of the executive management team in addressing quality system failures. Persistent issues such as the lack of science-based manufacturing process controls (process validation) and robust data-driven investigation programs have continued year after year. The warning letters issued during this period highlighted key 483 observations. The analysis reveals a positive correlation between observations related to the responsibilities of the quality unit and observations related to manufacturing equipment (design, suitability, cleaning, and maintenance). It indicates the importance for organizations to wholly adopt ASTM E25008 standards for manufacturing equipment design and verification. This would necessitate organizations to have sufficiently qualified quality unit team members capable of determining the design adequacies, allowing early involvement and implementation of design assessment programs assuring equipment/systems are indeed fit for the intended drug product and manufacturing process. The inspection data set highlights the ongoing challenges and the need for sustained improvements in the pharmaceutical manufacturing sector to ensure compliance and maintain high standards of patient safety and product quality.
References
- FDA Form 483 Frequently Asked Questions https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/inspection-references/fda-form-483-frequently-asked-questions
- Key Post-Pandemic Trends In Global FDA Observations For Drug Facilitie https://www.outsourcedpharma.com/doc/key-post-pandemic-trends-in-global-fda-observations-for-drug-facilities-0001#
- FDA Data Dashboard https://datadashboard.fda.gov/ora/index.htm
- Drug Establishments Current Registration Site https://www.fda.gov/drugs/drug-approvals-and-databases/drug-establishments-current-registration-site
- Warning Letters https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/compliance-actions-and-activities/warning-letters
- US FDA to boost inspections of drug manufacturing units in Indi https://www.reuters.com/business/healthcare-pharmaceuticals/us-fda-boost-inspections-drug-manufacturing-units-india-2024-02-27/
- The geography of prescription pharmaceuticals supplied to the USA: levels, trends, and implications https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8109232/pdf/lsaa085.pdf.
- ASTM E2500-20: Standard Guide for Specification, Design, and Verification of Pharmaceutical and Biopharmaceutical Manufacturing Systems and Equipment https://www.astm.org/e2500-20.html
About The Authors:
 Ajay Pazhayattil, Ph.D., is a seasoned management consultant and an industrial pharmacist with experience in the industry’s solid oral, sterile, and API sectors. He is the founder of cGMP World and a founding partner at Itaan Pharma, an injectable manufacturer. He has been in leadership roles with North American pharmaceutical brands, generics, and CDMOs, including VP of QA, scientific, and regulatory affairs at Capcium; quality director at Eurofins; and associate director at Apotex. Pazhayattil plays a key role in assisting organizations to navigate compliance and remediation efforts arising from FDA 483s and warning letter scenarios. He has been the lead author and contributor for industry guidance documents, including PDA, ISPE, AAPS, and RAPS.
Ajay Pazhayattil, Ph.D., is a seasoned management consultant and an industrial pharmacist with experience in the industry’s solid oral, sterile, and API sectors. He is the founder of cGMP World and a founding partner at Itaan Pharma, an injectable manufacturer. He has been in leadership roles with North American pharmaceutical brands, generics, and CDMOs, including VP of QA, scientific, and regulatory affairs at Capcium; quality director at Eurofins; and associate director at Apotex. Pazhayattil plays a key role in assisting organizations to navigate compliance and remediation efforts arising from FDA 483s and warning letter scenarios. He has been the lead author and contributor for industry guidance documents, including PDA, ISPE, AAPS, and RAPS.
 Satish Joshi is a consultant in the pharmaceutical industry with over 38 years of experience. He is the founder of SHJ Consulting Services, providing GMP consulting services. Having held senior leadership roles, including senior vice president of quality assurance at Hospira, Orchid Healthcare; as VP of quality, Dr. Reddy’s; and at Lupin, Satish has worked in functions such as QA, compliance, QC, analytical development, and packing development. His expertise extends to successfully navigating regulatory inspections from agencies such as U.S. FDA, MHRA, TGA, ANVISA, Taiwan, and GCC. Joshi is recognized for his pivotal role in setting up greenfield projects and establishing robust quality procedures and systems.
Satish Joshi is a consultant in the pharmaceutical industry with over 38 years of experience. He is the founder of SHJ Consulting Services, providing GMP consulting services. Having held senior leadership roles, including senior vice president of quality assurance at Hospira, Orchid Healthcare; as VP of quality, Dr. Reddy’s; and at Lupin, Satish has worked in functions such as QA, compliance, QC, analytical development, and packing development. His expertise extends to successfully navigating regulatory inspections from agencies such as U.S. FDA, MHRA, TGA, ANVISA, Taiwan, and GCC. Joshi is recognized for his pivotal role in setting up greenfield projects and establishing robust quality procedures and systems.
 Marzena Ingram is an independent senior pharmaceutical consultant with a wealth of experience in quality and technical operations and process validation. At the forefront of her responsibilities is the critical task of addressing FDA warning letter scenarios for clients, ensuring compliance and regulatory adherence. Her strategic leadership and attention to detail have enabled her to spearhead compliance programs and initiatives that meet global regulatory requirements and to set new benchmarks in the industry. She has developed and led specialized teams at high-volume manufacturing organizations. Ingram serves as the VP of ISPE Canada.
Marzena Ingram is an independent senior pharmaceutical consultant with a wealth of experience in quality and technical operations and process validation. At the forefront of her responsibilities is the critical task of addressing FDA warning letter scenarios for clients, ensuring compliance and regulatory adherence. Her strategic leadership and attention to detail have enabled her to spearhead compliance programs and initiatives that meet global regulatory requirements and to set new benchmarks in the industry. She has developed and led specialized teams at high-volume manufacturing organizations. Ingram serves as the VP of ISPE Canada.